Wyszukaj egzamin lub pytanie
Egzamin PES Genetyka kliniczna / jesień 2008
120 pytań
Pytanie 1
Odsetek wszystkich kolejnych raków piersi spowodowanych mutacją BRCA1 wynosi w Polsce około:
Pytanie 2
Zaznacz wskazania do wykonywania testów BRCA1:
1) każda chora z rakiem piersi;
2) każda chora z rakiem jajnika;
3) każda chora z rakiem trzonu macicy;
4) kobieta zdrowa z rakiem jajnika u babci ze strony ojca;
5) kobieta zdrowa z rakiem jajnika u siostry matki.
Prawidłowa odpowiedź to:
Pytanie 3
Liczba Polek - nosicielek mutacji BRCA1 u których w rodzinie nie stwierdzono zachorowań na raki piersi lub jajnika wynosi około:
Pytanie 4
„HNPCC susp.” Rozpoznasz wówczas gdy u kobiety 55-letniej z rakiem trzonu macicy stwierdzono:
1) drugi pierwotny nowotwór - raka piersi w wieku 50 lat;
2) drugi pierwotny nowotwór - raka jelita grubego w wieku 49 lat;
3) raka trzonu macicy u matki w wieku 45 lat;
4) raka jelita grubego u ojca w wieku 49 lat;
5) raka jelita grubego u babci ze strony matki w wieku 45 lat.
Prawidłowa odpowiedź to:
Pytanie 5
Zaznacz opcje terapeutyczne do rozważenia w leczeniu raka piersi u nosicielki mutacji BRCA1:
1) adnexektomia; 4) tamoxifen mimo ER(-) w guzie;
2) hysterektomia; 5) chemioterapia bez taksanów.
3) kolektomia;
Prawidłowa odpowiedź to:
Pytanie 6
Profilaktyka raka piersi/jajnika u nosicielki mutacji BRCA1 obejmuje takie działania jak:
1) doustna antykoncepcja powyżej 30 r.ż.; 4) adneksektomia;
2) długie karmienie piersią; 5) kolektomia.
3) tamoxifen;
Prawidłowa odpowiedź to:
Pytanie 7
Czułość mammografii w wykrywaniu wczesnego raka piersi u nosicielek mutacji BRCA1 wynosi około:
Pytanie 8
Kompletne remisje w terapii neoadjuwantowej raka piersi u nosicielek mutacji BRCA1 uzyskuje się poprzez:
Pytanie 9
Do charakterystycznych cech raków piersi u nosicielek mutacji BRCA1 należą:
1) histopatologiczny typ rdzeniasty; 4) HER 3+;
2) histopatologiczny typ zrazikowy; 5) obustronność.
3) brak receptorów estrogenowych w guzie;
Prawidłowa odpowiedź to:
Pytanie 10
Najczęstszą przyczyną zgonów pacjentek z rakiem piersi będących nosicielkami mutacji BRCA1 jest:
Pytanie 11
Zespół Lyncha charakteryzują:
1) mutacje STK 11;
2) mutacje MSH2;
3) zwiększona predyspozycja do raka jelita grubego;
4) zwiększona predyspozycja do raka trzonu macicy;
5) zwiększona predyspozycja do białaczki.
Prawidłowa odpowiedź to:
Pytanie 12
Mutacje w genie APC są przyczyną:
Pytanie 13
W rodzinie wykryto polipowatość jelita u nosiciela mutacji genu MYH. Zwiększone ryzyko powtórzenia się polipowatości do blisko 25% występuje u:
1) ojca; 2) matki; 3) braci; 4) sióstr; 5) dzieci.
Prawidłowa odpowiedź to:
Pytanie 14
Zaznacz cechy charakterystyczne rodzinnej polipowatości gruczolakowatej:
1) chorują tylko mężczyźni; 4) mutacje MSH2;
2) wysokie ryzyko raka jelita grubego przed 50 r.ż.; 5) mutacje APC.
3) wysokie ryzyko raka piersi powyżej 50 r.ż.;
Prawidłowa odpowiedź to:
Pytanie 15
W przebiegu VHL charakterystycznie występuje:
1) onkocytoma; 4) pheochromocytoma (guz chromo
2) guz ELST; chłonny nadnerczy);
3) hemangioblastoma móżdżku; 5) wysypka skórna.
Prawidłowa odpowiedź to:
Pytanie 16
Jedno z rodziców jest nosicielem translokacji chromosomowej wzajemnej. Przy określaniu prawdopodobieństwa urodzenia dziecka z niezrównoważonym kariotypem prowadzącym do wad rozwojowych dla tej pary należy rozważyć poniższe czynniki, z wyjątkiem:
Pytanie 17
Konsekwencjami klinicznymi nosicielstwa translokacji chromosomowych wzajemnych nie są:
Pytanie 18
Miarą prawdopodobieństwa urodzenia dziecka z niezrównoważonym kariotypem w rodzinach z nosicielstwem translokacji chromosomowych wzajemnych jest wskaźnik, który oznacza stosunek:
Pytanie 19
Prawdopodobieństwo urodzenia dziecka z niezrównoważonym kariotypem w rodzinie nosicieli translokacji chromosomowej wzajemnej jest niskie. Ryzyko poronień samoistnych w tej rodzinie jest:
Pytanie 20
Translokacje ryzyka podwójnego niezrównoważenia segmentu to:
Pytanie 21
Zapłodnienie gamet z chromosomami mejotycznymi powstałymi u nosicieli translokacji chromosomowych wzajemnych w wyniku rozdziału naprzemiennego, powoduje powstanie:
Pytanie 22
Rozpoznania cytogenetyczne zespołu Edwardsa:
Pytanie 23
Translokacja chromosomowa wzajemna zrównoważona nie jest czynnikiem ryzyka:
Pytanie 24
Wystąpienie fenotypu zespołu Patau (u dziecka pre- lub postnatalnie) jest prawdopodobne w rodzinie nosicieli następujących translokacji:
Pytanie 25
Zespół niewrażliwości na androgeny jest jednostką:
Pytanie 26
Gen ATM ulegający mutacjom a Ataksja Teleangiectasia koduje białko typu:
Pytanie 27
Ryzyko wystąpienia choroby dziedziczonej wieloczynnikowo u krewnych
I-go stopnia osoby chorej może być wyliczone w przybliżeniu z wzoru:
(f - oznacza częstość populacyjną choroby, h - dziedziczność choroby)
Pytanie 28
Pewne postacie złośliwej hypertermii zależą od defektu białka kanału wapniowego (receptora ryanodyny - toksycznego alkaloidu). Defekt taki powoduje śmiertelnie niebezpieczną nadwrażliwość na:
Pytanie 29
Zespół nagich limfocytów (BLS) jest przyczyną wczesnej śmierci dzieci z powodu wielkiej podatności na zakażenia. Przyczyną choroby może być:
Pytanie 30
Dlaczego zespoły wad wrodzonych uwarunkowane mutacjami genów homeoboksowych HOX (I klasa) i nie-HOX (II klasa) są rzadko spotykane u pacjentów zgłaszających się do poradni genetycznych?
Pytanie 31
Konieczność badania kariotypu dziecka z ewidentnym fenotypem zespołu Downa wynika z:
Pytanie 32
Badaniem diagnostycznym pozwalającym na rozpoznanie ponad 90% przypadków zespołu Pradera-Williego jest badanie:
Pytanie 33
Skutkiem disomii jednorodzicielskiej (UPD) może być:
Pytanie 34
Skutkiem nosicielstwa zrównoważonej translokacji wzajemnej może być:
Pytanie 35
Stopień penetracji genu to:
Pytanie 36
Nosicielstwo genetyczne:
Pytanie 37
Obligatoryjną nosicielką mutacji sprzężonej z chromosomem X jest kobieta spełniająca jedno z poniższych kryteriów:
Pytanie 38
Test predykcyjny (przedobjawowy) w kierunku mutacji genu IT15 powodującej bardzo wysoką predyspozycję do zachorowania powinno się wykonywać, wg obecnie obowiązujących zasad:
Pytanie 39
W poradni genetycznej konsultowane jest dziecko niskiego wzrostu, o prawidłowym rozwoju psychoruchowym, z niedokrwistością i małopłytkowością, z zaburzeniami rozwoju promieniowej strony kończyn górnych. Jakie rozpoznania należy wziąć pod uwagę i jakie badania genetyczne wykonać w ramach diagnostyki różnicowej?
Pytanie 40
Badania nabytych zmian genetycznych w nowotworach układu krwiotwórczego są przydatne w:
Pytanie 41
Chromosom Filadelfia (Ph):
Pytanie 42
Prawidłowe wartości poziomu glukozy we krwi noworodka urodzonego przedwcześnie wynoszą:
Pytanie 43
U chłopca 3-letniego, z opóźnieniem psychoruchowym, hipotonią mięśniową i pomarańczowym zabarwieniem zmoczonych pieluszek w okresie niemowlęcym, stwierdza się pourazowe ubytki warg i palców, okresy pobudzenia i niepokoju. Badanie molekularne wykazało mutację w genie HGRT:
1) wskaźnik kwasu moczowego do kreatyniny w moczu wynosi < 1;
2) wskaźnik kwasu moczowego do kreatyniny w moczu wynosi > 1;
3) poziom kwasu moczowego w surowicy przekracza 8 mg%;
4) w moczu obecne są liczne erytrocyty;
5) podobną chorobę stwierdzono u starszej siostry pacjenta.
Prawidłowa odpowiedź to:
Pytanie 44
U noworodka urodzonego o czasie, w dobrym stanie ogólnym, karmionego piersią, w trzecim dniu życia, bez wyraźnej przyczyny, doszło do szybkiego pogorszenia stanu ogólnego. Pojawiły się: niechęć do ssania, ulewania, senność przechodząca w śpiączkę, drżenia kończyn, obniżenie napięcia mięśni. Wywiad rodzinny, ciążowy i okołoporodowy bez obciążeń. Równocześnie z rutynowym postępowaniem neonatologicznym i diagnostyką w kierunku zakażeń, należy pilnie wysłać materiał do odpowiednich laboratoriów pediatrii metabolicznej celem wykonania a/ analizy profilu kwasów organicznych w jednorazowej porcji moczu metodą GC-MS i b/ analizy aminokwasów i acylokarnityn w suchej kropli krwi pobranej na bibułę metodą tandemowej MS. Analizy te pozwolą na rozpoznanie lub wykluczenie następujących wad metabolizmu:
Pytanie 45
U chłopca 3-letniego, dotychczas pozornie zdrowego, wystąpiły dolegliwości bólowe w czasie chodzenia i krzywicze zniekształcenie kończyn mimo podawania profilaktycznego witaminy D zgodnie z zalecanymi schematami. Starsza siostra zmarła w okresie niemowlęcym z powodu krwawień i narastającej marskości wątroby bez ustalonego rozpoznania. Skrining selektywny w kierunku wad metabolizmu pozwolił na rozpoznanie tyrozynemii typu I. Które z poniższych stwierdzeń jest prawdziwe?
1) rozpoznanie ostateczne ustalono na podstawie zwiększonego wydalania w moczu bursztynyloacetonu;
2) rozpoznanie ostateczne ustalono na podstawie zwiększonego stężenia tyrozyny w osoczu;
3) rozpoznanie ustalono poprzez analizę profilu aminokwasów i acylokarnityn w suchej kropli krwi metodą tandemowej MS;
4) rozpoznanie ustalono poprzez analizę profilu kwasów organicznych w moczu metodą GC-MS;
5) rozpoznanie ustalono na podstawie analizy ilościowej aminokwasów w osoczu (aminoacydogram).
Prawidłowa odpowiedź to:
Pytanie 46
Epizody „intoksykacji” endogennej z uszkodzeniem funkcji wątroby mogą występować w przebiegu wielu wad metabolizmu, w tym w:
1) zaburzeniach beta-oksydacji kwasów tłuszczowych; 4) acyduriach organicznych;
2) hiperamonemach wrodzonych; 5) tyrozynemii typu I.
3) deficycie biotynidazy;
Prawidłowa odpowiedź to:
Pytanie 47
Obecnie znamy skuteczne leczenie przyczynowe dla następujących genetycznych wad metabolizmu:
1) galaktozemia; 4) choroba Gauchera;
2) tyrozynemia typu I; 5) zaburzenia beta-oksydacji kwasów
3) choroba Lesch-Nyhana; tłuszczowych.
Prawidłowa odpowiedź to:
Pytanie 48
Przy założeniu, że definicja choroby mitochondrialnej jest następująca: „choroba uwarunkowana pierwotnym zaburzeniem funkcji łańcucha oddechowego” wskaż możliwe sposoby dziedziczenia tych chorób:
1) mitochondrialne (matczyne); 4) sprzężone z chromosomem X;
2) autosomalne recesywne; 5) sporadyczne występowanie.
3) autosomalne dominujące;
Prawidłowa odpowiedź to:
Pytanie 49
Przyczyną powiększenia i/lub uszkodzenia funkcji wątroby u niemowlęcia mogą być liczne wrodzone wady metabolizmu, w tym galaktozemia, wrodzona nietolerancja fruktozy, tyrozynemia typu I, choroby lizosomalne, glikogenozy, choroba Wilsona, deficyt alfa-1-antytrypsyny, zaburzenia glikozylacji białek, zaburzenia syntezy kwasów tłuszczowych, choroby peroksysomalne, choroby mitochondrialne (deplecje mtDNA), hiperamonemie pierwotne i inne. Niektóre z nich można wstępnie rozpoznawać na poziomie ogólnopediatrycznym, na podstawie wywiadu, badania przedmiotowego, podstawowych badań biochemicznych i obserwacji klinicznej. Które z wymienionych chorób mogą być zdiagnozowane na miejscu, poprzez przesłanie materiału do odpowiednich krajowych laboratoriów metabolicznych i nie wymagają hospitalizacji w ośrodku pediatrii metabolicznej?
1) wrodzona nietolerancja fruktozy, galaktozemia, deficyt alfa-1-antytrypsyny;
2) choroby lizosomalne i peroksysomalne, zaburzenia glikozylacji białek;
3) tyrozynemia typu I, hiperamonemie;
4) choroby mitochondrialne, zaburzenia syntezy kwasów tłuszczowych;
5) choroba Wilsona, glikogenozy.
Prawidłowa odpowiedź to:
Pytanie 50
U 3-miesięcznego niemowlęcia, dotychczas zdrowego, po podaniu szczepionki (zgodnie z kalendarzem szczepień) doszło do gwałtownego pogorszenia. W ciągu kilku godzin rozwinęły się objawy przypominające zapalenie mózgu, zatrucie chemiczne lub zespól Reye’a: brak łaknienia, wymioty, cechy uszkodzenia wątroby, żółtaczka, utrata kontaktu, drgawki, śpiączka. W ciągu 3 dni dziecko zmarło mimo intensywnego leczenia objawowego i przeciwzapalnego bez ustalonego rozpoznania. Na jakiej podstawie wśród niżej wymienionych można pośmiertnie rozpoznać lub wykluczyć genetycznie uwarunkowane zaburzenia beta-oksydacji kwasów tłuszczowych?
1) pośmiertne badanie makroskopowe;
2) pośmiertne badanie histologiczne;
3) pośmiertne badanie mikroskopowo-elektronowe;
3) analiza metodą GC-MS zamrożonej próbki moczu z początkowego okresu choroby;
4) analiza acylokarnityn w suchej kropli krwi metodą tandemowej MS z początkowego okresu choroby;
5) próbka moczu, surowicy i skk, pobrane bezpośrednio przed zgonem.
Prawidłowa odpowiedź to:
Pytanie 51
Według europejskiej definicji, „choroba rzadka” (rare disease) to stan chorobowy występujący w populacji z częstością mniejszą niż:
Pytanie 52
Które z wymienionych poniżej zaburzeń metabolicznych i genetycznych należą do chorób rzadkich według nomenklatury europejskiej?
Pytanie 53
Które z wymienionych poniżej objawów klinicznych charakteryzują zespół wrodzonych zaburzeń glikozylacji typu Ib (CDG Ib)?
1) uporczywe biegunki;
2) postępujące uszkodzenie wątroby;
3) wysięk w osierdziu;
4) opóźnienie rozwoju psychoruchowego i drgawki;
5) nieprawidłowy wzór glikozylacji transferyny w surowicy.
Prawidłowa odpowiedź to:
Pytanie 54
Typową wadą dla zespołu Patau’a jest:
Pytanie 55
Charakterystyczną cechą dotyczącą dziedziczenia achondroplazji jest:
Pytanie 56
Przyczyną zespołu Angelmana może być:
Pytanie 57
Jednym z kryteriów rozpoznania choroby von Recklinhausena jest:
Pytanie 58
Badanie, które należy wykonać co dwa lata przy podejrzeniu mutacji genów mutatorowych to:
Pytanie 59
Odstępstwa od dziedziczenia mendlowskiego dotyczące zespołu łamliwego chromosomu X fra(X) przejawiają się w:
Pytanie 60
Charakterystycznym objawem zespołu Waardenburga jest:
Pytanie 61
Wczesnymi objawami dystrofii mięśniowej Duchenne’a są:
Pytanie 62
Wskaż zdanie prawdziwe dotyczące różnicowania płci męskiej:
Pytanie 63
Wskaż zdanie prawdziwe dotyczące rejonów pseudoautosomalnych chromosomów X i Y:
Pytanie 64
Do charakterystycznych objawów choroby Huntingtona dla postaci młodzieńczej należy:
Pytanie 65
Mutacje w genie RET mogą być odpowiedzialne za rozwój:
Pytanie 66
Które z poniższych chorób o podłożu dziedzicznym są przekazywane zgodnie z prawami Mendla?
Pytanie 67
Za nowotwory dziedziczne najczęściej odpowiedzialne są geny:
Pytanie 68
Stałą cechą fenotypu niemozaikowego zespołu Klinefeltera jest:
Pytanie 69
Wskaż zdanie prawdziwe dotyczące rozwoju macicy u kobiet z kompletną postacią zespołu niewrażliwości na androgeny:
Pytanie 70
Wskaż zdanie prawdziwe dotyczące zagłębienia w okolicy krzyżowej:
Pytanie 71
Oznaczenie markera Ca125 w surowicy krwi zlecisz u nosicielki mutacji w genie:
Pytanie 72
Która z wymienionych zygot zawiera materiał genetyczny który prowadzi w 100% do poronienia lub wczesnego zgonu noworodka?
Pytanie 73
Analiza rodowodu wskazuje, że choroba przekazywana jest pionowo w kolejnych, pokoleniach, mniej więcej z równą liczbą chorych kobiet i mężczyzn. Jest też przekazywanie choroby z ojca na syna.
Przedstawiony rodowód wskazuje, że jest to dziedziczenie:
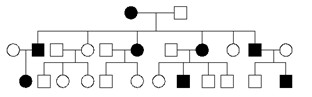
Pytanie 74
Stwierdzenie tylko „bruzdy poprzecznej na jednej dłoni” najbardziej wskazuje na:
Pytanie 75
Trzy małe anomalie:
Pytanie 76
Które ze stwierdzeń dotyczących choroby Huntingtona nie jest prawdziwe?
Pytanie 77
Choroba o różnym początku i przebiegu, powodująca między innymi - osłabienie mięśni, kardiomiopatię, kwasicę mleczanową oraz napady padaczkowe i udarowe dotyczy kilku osób w przedstawionym rodowodzie. Jaki jest najbardziej prawdopodobny tryb dziedziczenia w/w choroby i które zjawisko najlepiej objaśnia zmienną ekspresję objawów oraz jakie przyjmuje się ryzyko powtórzenia choroby (rodowód w tym wypadku służy jedynie do ułatwienia interpretacji sposobu przekazywania)?
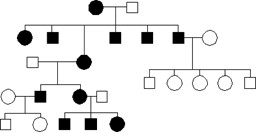
Pytanie 78
Ryzyko powtórzenia się wrodzonej i izolowanej wady rozwojowej o uwarunkowaniu wieloczynnikowym:
1) zmniejsza się po każdej prawidłowej ciąży zakończonej urodzeniem zdrowego dziecka;
2) zwiększa się z każdym kolejnym przypadkiem urodzenia dziecka z podobną ciężką izolowaną wrodzoną wadą rozwojową, bez względu na kolejność ciąży;
3) jest stałe oparte na obserwacjach w danej populacji i dla każdej wady wynosi ok. 5%;
4) zwiększa się w każdym kolejnym pokoleniu;
5) jest mniejsze im większa jest ekspresja wady i gdy dotyczy to płci mniej oczekiwanej.
Prawidłowa odpowiedź to:
Pytanie 79
Konsultujący genetyk został poproszony do 3 tygodniowego noworodka przyjętego głównie z powodu wymiotów oraz z obojnaczymi narządami płciowymi. Podstawowe badania wykazały zaburzenia elektrolitowe - niskie wartości jonów Na+ oraz lekko podwyższone jony K+. Przeprowadzone badania wykazały podwyższone wartości keto steroidów oraz pregnandiolu w moczu, a w surowicy krwi znacznie podwyższone stężenia 17α-hydroksyprogesteronu oraz ACTH. Jaka może to być jednostka chorobowa i jej domniemana przyczyna oraz jak jest ona uwarunkowana?
Pytanie 80
Do poradni genetycznej zgłaszają się rodzice z dzieckiem płci męskiej w wieku wczesnoniemowlęcym, z parametrami rozwoju fizycznego - zwłaszcza masą ciała i długością > 97 centyla oraz z dość muskularną, budowa wskazująca na znacznie starsza dziecko. Chłopczyk jest po zabiegu przepukliny sznura pępowinowego, nieprawidłowym napieciem mięśni, cechami dysmorfii głownie w obrębie twarzy w tym lekka asymetria, znaczne powiększenie języka, płaski naczyniak okolicy glabelli, a na płatkach małżowin usznych skośne bruzdowane linie. Palpacyjnie wątroba wystaje poniżej łuku żebrowego na 2-3 centymetrów, po badaniu USG stwierdzono także dość duże nerki. Cechy kliniczne pozwoliły na wysunięcie podejrzenia zespołu:
1) Beckwith-Wiedemanna; 4) Perlmana;
2) Simpson - Golabi - Behmel; 5) Pallister - Killian.
3) Sotosa;
Które z tych zaburzeń może manifestować się połowiczym przerostem ciała, zwiększonym ryzykiem rozwoju guza Wilmsa oraz być spowodowane disomią ojcowską chromosomu 11?
Pytanie 81
Jaka to choroba, która charakteryzuję się występowaniem już w okresie noworodkowym: uporczywych wymiotów, biegunką, żółtaczką, powiększeniem i narastającą niewydolnością wątroby, brakiem przyrostu masy ciała, obuoczną zaćmą oraz skłonnościami do infekcji - nawet posocznicy wywołanej przez bakterie Gram-ujemne. Jaki jest sposób jej dziedziczenia i niedobór/brak aktywności, którego z enzymów warunkujących tą chorobę stwierdzany jest najczęściej i co służy do badania?
Pytanie 82
Który z tzw. zespołów genów sąsiadujących (contiguous gene syndrome) charakteryzuje się między innymi: specyficznym wyglądem twarzy określanym jako „twarz Elfa”, charakterystyczną osobowością ocenianą jako „cocktail party”, zaburzeniami poznawania przestrzenno-wzrokowego oraz nadzastawkowym zwężeniem aorty (SVAS). Jaki rodzaj zaburzenia jest przyczyną tego zespołu i niedobór produktu jakiego genu jest odpowiedzialny za wadę SVAS i innych tkanek strukturalnych?
Pytanie 83
Genetyk kliniczny został poproszony o konsultację noworodka u którego stwierdzono: hipotrofię wewnątrzmaciczną, znaczne obniżenie napięcia mięśniowego, szczątkową heksodaktylię pozaosiową obu rąk, u stóp częściową syndaktylię palców 2 i 3 w kształcie litery Y, wnętrostwo i spodziectwo, ponadto cechy dysmorfii w obrębie głowy i twarzy - małogłowie, zwężenie dwuskroniowe, rozszczep podniebienia wtórnego, krótki zadarty nosek z płaską nasadą, zmarszczki nakątne, skośno-dolnie ustawione szpary powiekowe i ptozę. W narządach wewnętrznych stwierdzono wadę serca (wspólny kanał przedsionkowo-komorowy), wodonercze, hipoplastyczne ciało modzelowate. Jaki wrodzony zespół wad rozwojowych charakteryzuje ten opis, jaki gen jest głównie odpowiedzialny za jego powstanie i w jakim szlaku sygnałowym bierze udział produkt pośredni jego genu i jaki jest sposób dziedziczenia?
Pytanie 84
17-letnia pacjentka z pierwotnym brakiem miesiączki. W badaniu przedmiotowym stwierdzono wzrost na poziomie 75 centyla, prawidłowy rozwój zewnętrznych narządów płciowych, brak owłosienia pachowego i łonowego, rozwój gruczołów piersiowych. Badania hormonalne pozwoliły na wykazanie między innymi prawidłowych poziomów gonadotropin. Najbardziej prawdopodobnym rozpoznaniem jest u niej:
Pytanie 85
Z podanych poniżej rozpoznań cytogenetycznych wskaż to, które odbiega swoim charakterem od pozostałych:
Pytanie 86
Która z podanych niżej mutacji może wywołać potencjalne najpoważniejsze (nawet letalne) skutki?
Pytanie 87
Matka urodziła z ciąży I syna z klinicznymi cechami zespołu Downa i kariotypem 46,XY,der(14;21)q10;q10),+21. Po przeprowadzeniu oceny kariotypu u niej i u jej męża okazało się, że pacjentka ta jest nosicielką fuzji centrycznej między chromosomami 14 i 21 [kariotyp 45,XX,der(14;21)(q10;q10)]. Jakie jest teoretyczne prawdopodobieństwo wystąpienia w jej komórce jajowej składu chro-mosomów warunkującego po zapłodnieniu nieprawidłowości rozwojowe u płodu?
Pytanie 88
U noworodka z podejrzeniem wrodzonego przerostu kory nadnerczy stwierdzono kariotyp 46,XX oraz maskulinizację zewnętrznych narządów odpowiadającą V stopniowi w skali Pradera. W mosznie nie wyczuwano tworów mogących odpowiadać gonadom. Ten obraz kliniczny może odpowiadać cechom:
Pytanie 89
Podczas badań przesiewowych w 13 tygodniu pierwszej ciąży 25-letniej ciężarnej stwierdzono przezierność karkową (NT) równą 4 mm, podwyższony poziom podjednostki beta wolnej gonadotropiny łożyskowej (fβhCG) i obniżony poziom osoczowego białka ciążowego A (PAPP-A). Parametry takie mogą przemawiać za:
1) brakiem nieprawidłowości rozwojowych płodu na tym etapie ciąży;
2) podejrzeniem u płodu zespołu Downa;
3) podejrzeniem u płodu zespołu Turnera;
4) podejrzeniem u płodu triploidii pochodzenia matczynego;
5) podejrzeniem u płodu zespołu Edwardsa.
Prawidłowa odpowiedź to:
Pytanie 90
Rozwój raka jelita grubego jest przykładem wieloetapowości procesu karcinogenezy, z kumulacją szeregu zmian genomowych, wiążących się z kolejnymi stadiami rozwoju nowotworu od prawidłowego nabłonka, poprzez hiperplazję nabłonka, kolejne etapy gruczolakowatości błony śluzowej jelita (gruczolaki wczesne, pośrednie, późne) z rozwojem w finale raka i uzyskaniem przez niego zdolności do przerzutowania. Związek między kumulacją mutacji a stopniem zaawansowania karcinogenezy określa się torem mutacyjnym. Wśród czynników etiopatogenetycznych w raku jelita grubego wymienia się:
1) zjawisko hipometylacji DNA;
2) mutacje genu TP53 („strażnik genomu”), kodującego białko p53;
3) mutacje genu APC;
4) mutacje genu Ki-RAS;
5) mutacje genu DCC.
Wskaż, w którym z poniższych szeregów uporządkowano wymienione zmiany genomowe w sposób najlepiej odpowiadający torowi mutacyjnemu w raku jelita grubego:
Pytanie 91
Która z podanych niżej chorób lizosomalnych związana jest z procesem postępującej demielinizacji?
Pytanie 92
Który z podanych niżej markerów nie może być traktowany z formalnego punktu widzenia jako element przedurodzeniowej diagnostyki przesiewowej w I trymestrze ciąży?
Pytanie 93
Który z poniższych przykładów nie powinien być traktowany jako wskazanie do inwazyjnej diagnostyki przedurodzeniowej?
Pytanie 94
U 2-letniego chłopca stwierdzono lewostronne wnętrostwo. W teście chromatyny Y stwierdzono w obrębie jąder jego limfocytów krwi obwodowej 2 grudki chromatyny Y (Y-bodies). Na podstawie pełnej analizy cytogenetycznej rozpoznano u niego kariotyp 47,XYY.
U pacjenta przeprowadzono orchidopeksję. Które z poniższych stwierdzeń można uznać za właściwe z punktu widzenia informacji przekazywanych rodzicom o potencjalnych skutkach tej aberracji u syna?
Pytanie 95
Wśród najczęstszych przyczyn upośledzenia umysłowego na drugim miejscu można postawić zespół:
Pytanie 96
Para młodych, niespokrewnionych pacjentów została skierowana do Poradni Genetycznej w celu uzyskania porady. Ciąża pierwsza została zakończona cięciem cesarskim w 30 tygodniu. Noworodek płci żeńskiej wykazywał znaczną hipotonię, punktacja w skali Apgar w 1, 3 i 5 minucie życia - 3 punkty, w 10 minucie życia - 4 punkty. Dziecko zmarło w 3 dobie życia z powodu narastającej niewydolności oddechowej. Badanie USG wykonane w I trymestrze ciąży, nie wykazało istotnych odchyleń, stężenie α-fetoproteiny w surowicy krwi matki w normie. Badanie USG w II trymestrze wykazało wielowodzie, poza tym nie stwierdzono nieprawidłowości. Badanie echokardiograficzne płodu wykazało prawidłowy obraz serca płodu. Matka zgłaszała słabą wyczuwalność ruchów płodu.
U matki, w 15 roku życia stwierdzono migotanie przedsionków, złą tolerancję wysiłku, opadanie powiek, zaćmę, postępujące osłabienie dystalnych mięśni kończyn oraz zaniki mięśni mimicznych twarzy. Wywiad ojca bez istotnych obciążeń.
Najbardziej zasadne dalsze postępowanie to:
Pytanie 97
U 5-letniej pacjentki stwierdzono cienką, „prześwitującą” skórę, skłonność do tworzenia podbiegnięć krwawych, trudne gojenie się skaleczeń oraz zwiększoną ruchliwość w stawach ręki i w stawach łokciowych. Wskaż odpowiedź, w której prawidłowo podano najbardziej prawdopodobne rozpoznanie oraz informacje dotyczącą tej choroby:
Pytanie 98
Ze związku mężczyzny, chorego na hemofilię A oraz zdrowej kobiety urodziło się dziecko z zespołem Turnera o kariotypie 45,X. Dziecko wykazuje prawidłową aktywność czynnika VIII. U którego z rodziców doszło do nieprawidłowej segregacji chromosomów podczas mejozy?
Pytanie 99
Inwersję pericentryczną chromosomu 9, opisywaną jako przykład heteromorfizmu chromosomów należy opisać wg systemu ISCN 2005 jako:
Pytanie 100
Do Poradni Genetycznej zgłasza się 30-letnia kobieta ciężarna w 8 tygodniu ciąży w celu wykonania badań prenatalnych. Jej siostra urodziła 2 dni temu dziecko z zespołem Downa rozpoznanyn postnatalnie na podstawie obrazu klinicznego. Jakie postępowanie jest najwłaściwsze w opisywanej sytuacji?
Pytanie 101
Kariotyp zdrowej nosicielki fuzji centrycznej chromosomów 14 i 21, powinien być wg systemu ISCN 2005 zapisany jako:
Pytanie 102
Do chromosomów akrocentrycznych należą chromosomy par:
Pytanie 103
Delecja dwóch aminokwasów w łańcuchu polipeptydowym mogła pojawić się na skutek następującej mutacji:
Pytanie 104
Substytucja jednej pary zasad w eksonie może prowadzić do:
1) braku zmian w kodowanym łańcuchu polipeptydowym;
2) substytucji jednego aminokwasu w kodowanym łańcuchu peptydowym;
3) produkcji krótszego polipeptydu przez przedwczesne wystąpienie kodonu STOP;
4) uaktywnienie się ukrytego miejsca splicingu;
5) przesunięcia ramki odczytu w genie.
Prawidłowa odpowiedź to:
Pytanie 105
Wirusy, stosowane jako wektory do wprowadzania obcych sekwencji do komórek eukariotycznych najczęściej należą do:
Pytanie 106
Odmienność metody MLPA od innych metod opartych na reakcji PCR polega na tym, że:
Pytanie 107
Która z poniższych chorób jest determinowana ekspansją powtórzeń trinukleotydowych?
Pytanie 108
siRNA to:
Pytanie 109
Który z poniższych objawów nie zalicza się do „dużych” klinicznych kryteriów zespołu Marfana?
Pytanie 110
U noworodka stwierdza się zaburzone różnicowanie płci, IV° wg Klasyfikacji Pradera. Badaniem usg wykazano obecność macicy oraz jajników. Które z poniższych rozpoznań jest najbardziej prawdopodobne?
Pytanie 111
W którym z poniższych stanów istnieje przeciwwskazanie do stosowania Vit D3?
Pytanie 112
U 5-miesięcznego niemowlęcia stwierdza się rozsianą erytrodermię z pojawieniem się szkarlatynopodobnej wysypki w regionach zgięć stawowych i wokół naturalnych otworów ciała. Śluzówki jamy ustnej są wolne od zmian. Objaw Nikolskiego jest dodatni. Po 3 dniach obserwuje się płatowe łuszczenie naskórka, prowadzące do odsłonięcia wilgotnych, połyskujących regionów skóry. Jakie jest najbardziej prawdopodobne rozpoznanie?
Pytanie 113
Do poradni zgłaszają się rodzice z noworodkiem, u którego stwierdza się niedobór masy ciała (< 10 cnt), małogłowie (<10 cnt) oraz cechy dysmorfii twarzy pod postacią krótkich szpar powiekowych, ścieńczałej wargi górnej oraz wygładzenia rynienki nosowo-wargowej. U dziecka wykonano wcześniej kariotyp, który jest prawidłowy. Powyższy obraz wymaga ukierunkowania wywiadu od matki w odniesieniu do:
Pytanie 114
We wszystkich poniższych zespołach często można stwierdzić obecność wad wrodzonych serca. Połącz poniższe zespoły z najbardziej typowymi dla nich wadami:
1) zespół Downa a) ASD;
2) zespół Turnera b) COA;
3) zespół DiGeorge’a c) przerwanie łuku;
4) zespół Williamsa d) nadzastawkowe zwężenie aorty;
5) zespół Holta i Orama e) kanał przedsionkowo - komorowy.
Prawidłowa odpowiedź to:
Pytanie 115
Polidaktylia jest częstym objawem poniższych jednostek, z wyjątkiem:
Pytanie 116
Wskazaniem do przeprowadzenia prenatalnej diagnostyki przedurodzeniowej nie jest:
Pytanie 117
W konwencji przyjętej przez Komitet ds. Nomenklatury HUGO (Human Genome Organization), nazwa segmentu DNA: D3S2550E oznacza segment:
Pytanie 118
Niepełna penetracja etiopatogenetycznej mutacji oznacza, że:
Pytanie 119
Negatywnymi regulatorami cyklu komórkowego są:
Pytanie 120

